Research - projects
Current research topics

A. H2020-MSCA-ITN-2019, Νο 861046, BIOREMIA :
BIOfim-REsistant Materials for hard tissue Implant Applications:
PhD student: Yannick Fortouna (8-2020 until today),
Collaborations,
Publications

B. H2020-MSCA-ITN-2014, Νο 642642, SELECTA :
Smart ELECTrodeposited Alloys for environmentally sustainable applications:
from advanced protective coatings to micro/nano-robotic platforms
PhD student: Carla Cutrano (10/2015 - 5/2020),
Collaborations,
Publications

C. FP7-PEOPLE-2010-ITN, No 264635, BioTiNet :
Biocompatible Titanium-based Structures for Orthopaedics
PhD student: J.J. Gutierrez-Moreno (3/2011 - 11/2014) ,
Collaborations,
Publications
Other research topics
D. Bulk metallic glasses(UoI projects' responsible Prof. G.A.Evangelakis: VitriMetTech FP7-PEOPLE-2013-ITN, PlasticMetalGlass FP7-PEOPLE-ITN-2008)
PhD student: G. Bokas (1/2011 - 3/2014), Collaborations, Publications
E. Graphene or Carbon nanotubes' interactions with metals or metal oxides
PhD student: M. Gialampouki (12/2010 - 2/2014), Collaborations, Publications
F. Biological molecules (Flavonoids) and metallic ions (Fe, Cu, Zn) complexes
G. Metal nitrides
H. Metallic surfaces
I. SCIENCE IN SOCIETY

FP7-SiS-2008-1, No 230253, DIVERSITY
Improving the gender diversity management in materials research institutions.
A. BIOREMIA
BIOfilm-REsistant Materials for hard tissue Implant Applications
Project : H2020-MSCA-ITN-2019, Νο 861046 (2020-2023) http://www.bioremia.eu
BIOREMIA aims to develop innovative materials with enhanced antibacterial and antifouling functionality that will result in improved biological acceptance of implants for bone-related applications (orthopedics and dentistry). We will use state of the art materials and surface modification technologies designed to potentially limit the initial stages of bacterial adhesion, as well as biofilm formation.
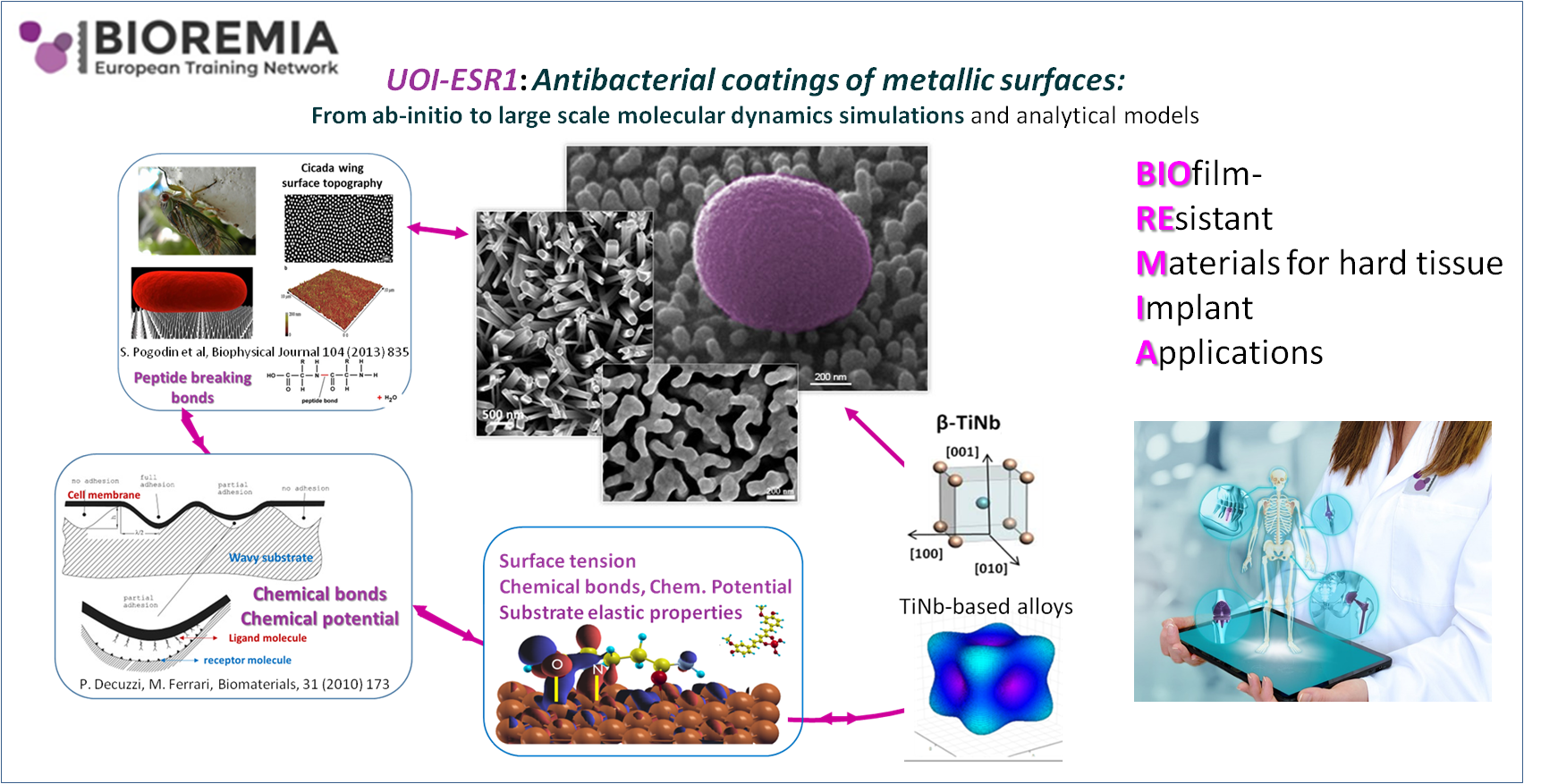
Figure : The UoI theoretical role.
B. Smart ELECTrodeposited Alloys for environmentally sustainable applications
from advanced protective coatings to micro/nano-robotic platforms
Project : H2020-MSCA-ITN-2014, Νο 642642 (2015-2018) http://selecta-etn.eu/project
SELECTA will explore new types of electrodeposited alloys (based on Fe, Cu or Al; free from hazardous and scarce raw elements), with tunable structure (amorphous, nanocrystalline) morphology (dense, nanoporous) and geometry (films, mocropillars, nanowires), to meet specific technological demands (high wear/corrosion resistance, superior magnetic properties or hydrophobicity) as written in part B along with training of young researchers that is the primary goal.
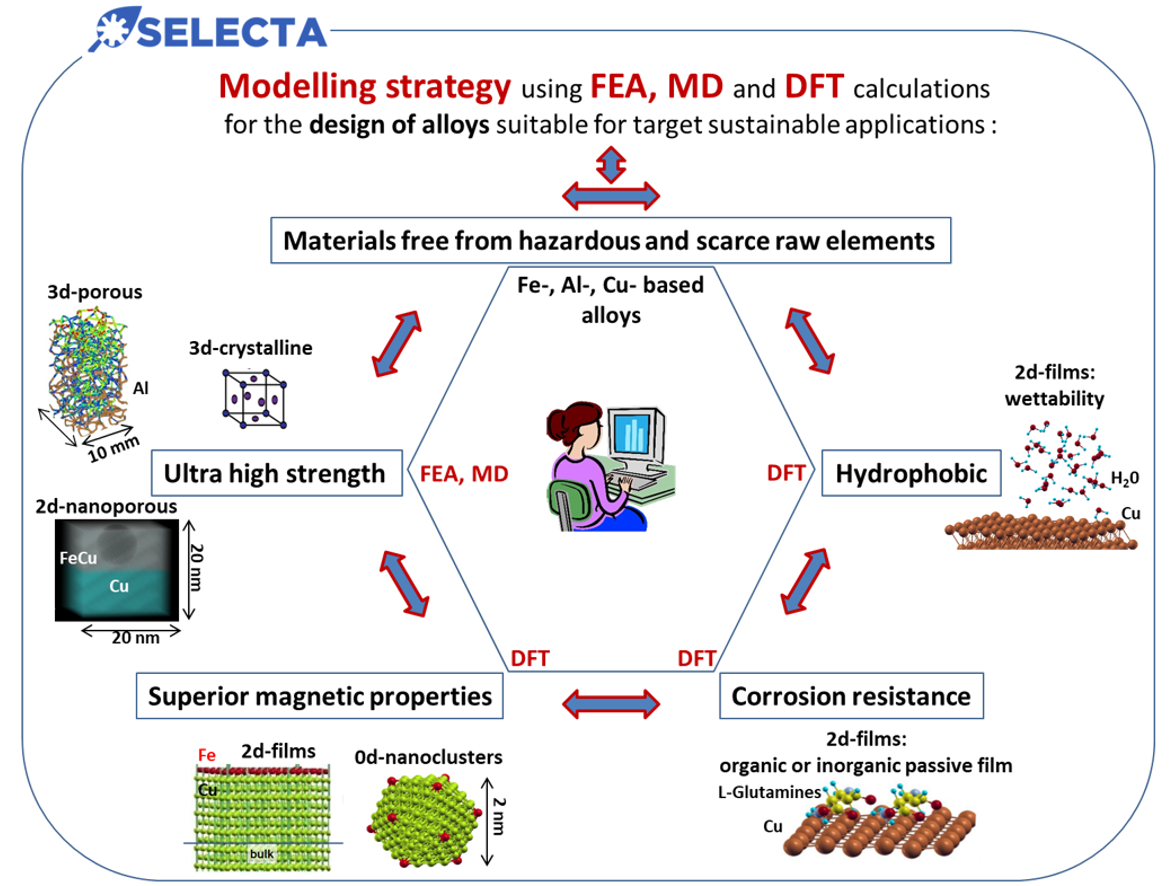
Figure : Illustration of the Modelling strategy in order to address SELECTA's requirements using Density Functional Theory (DFT), Molecular Dynamics (MD) and Finnite Element Analysis (FEA)

C. Biocompatible Titanium-based Structures for Orthopaedics
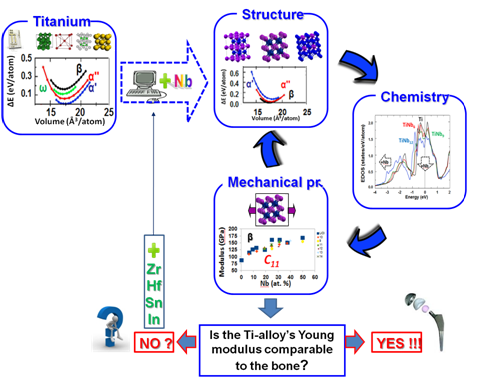
Project : FP7-PEOPLE-2010-ITN (# 264635), Academic-Industrial Initial Training Network on Innovative Biocompatible Titanium-based Structures for Orthopaedics (BioTiNet) 2011-2014 http://www.biotinet.eu/Theory-guided bottom-up design of low-rigidity Ti-based alloys
The project objective is to develop a computational procedure (from ab-initio towards large scale molecular
dynamics simulations) for the investigation and design of low rigidity Ti-based alloys. This project aims
in studying and understanding the relations between the mechanical properties and the structure and
chemistry of existing and/or proposed β-type Ti-based alloys, goal being the design of new β-type
Ti-based alloys. Tasks: a) physical and chemical insight of existing and/or proposed Ti-based systems
by means of the prediction of the basic thermodynamic, mechanical and chemical properties based on ab-initio
calculations for the crystalline alloys, b) Design of new Ti-based crystalline systems from ab-initio,
c) Scale-up to nanocrystalline via modeling of suitable semi-empirical approaches: from total energy
and ab-initio computations to interatomic model interactions and large scale parallel Computations and
Simulations and d) Simulation into real cases (e.g. β-type and nano-structured Ti-based, as well as
glassy alloys) and comparison with the other experimental data.
Collaboration:
Coordinator BioTiNet group: Prof. Mariana Calin, Dr. Annett Gebert and Prof. Jurgen Eckert, Institude of Complex Materials, IFW Dresden, Germany http://www.ifw-dresden.de/institutes/ikm/Other BioTiNet partners: Prof. Reza Yavary, Grenoble, France and Prof. Nicole Grobert, Oxford, UK
UoI BioTiNet group: Assist. Prof. Ch.E. Lekka, Accos. Prof. D.G. Papageorgiou and Prof. G.A.Evangelakis
PhD student
Jose Julio Guttierrez MorenoThesis: Theory-guided bottom-up design of low-rigidity Ti-based alloys (ab initio and molecular dynamics calculations)
http://cmsl.materials.uoi.gr/people.php/
Undergraduate student
Alexis PapaioannouThesis: Structural and electronic properties of Ti-xNb-Y (x=14:40, Y=Si,In, Sn) biocompatible alloys
http://cmsl.materials.uoi.gr/people.php/
Publications
D. Graphene or Carbon nanotube interactions with metals or metal oxides
D.1. Graphene growth on Cu substrates
Controlling the Orientation, Edge Geometry and Thickness of Chemical Vapor Depostion Graphene
A new way of growing graphene without the defects that weaken it and prevent electrons from flowing freely within it could open the way to large-scale manufacturing of graphene-based devices with applications in fields such as electronics, energy, and healthcare.
A team led by Nicole Grobert has overcome a key problem of growing graphene by chemical vapour deposition. by using copper substrate to control the orientation of growing graphene flakes. Where these flakes, called ‘domains’, are well-aligned, which will create a neater, stronger, and more ‘electron-friendly’ material.
The Oxford-led team, which includes researchers from Forschungszentrum Juelich Germany, the University of Ioannina Greece, and Renishaw plc, has also shown that it is also possible using the new technique to selectively grow bilayer domains of graphene – a double layer of closely packed carbon atoms – which are of particular interest for their unusual electrical properties.
http://www.materials.ox.ac.uk/blog/148/544/Controlling-the-Orientation-Edge-Geometry-and-Thickness-of-Chemical-Vapor-Depostion-Graphene.html
The UoI contribution:
The orientation of the graphene flakes along e.g. the Cu(110) surface <101> direction were supported by Density Functional Theory calculations revealing that the zig-zag edge is preferentially aligned along the surface channel due to Cu3d- C2p hybridizations, thus reducing the lattice mismatch.

Collaboration:
Prof. Nicole Grobert, Department of Materials, University of Oxfordhttp://www.materials.ox.ac.uk/peoplepages/grobert.html
Publications
Murdock, A.T., Koos, A., Britton, T.B., Houben, L., Batten, T., Zhang, T., Wilkinson, A.J., Dunin-Borkowski, R.E., Lekka, C.E., Grobert, N.
ACS Nano 7 (2) (2013) pp. 1351-1359
DOI: 10.1021/nn3049297
MsD student
Athanasia-Kiriaki MpalermpaThesis: "Structural and transport properties of Graphene flakes on Cu(110) surface"
http://cmsl.materials.uoi.gr/people.php/
Undergraduate student
Alexis KotanidisThesis: "Structural properties of Graphene flakes on Cu(001) surface"
http://cmsl.materials.uoi.gr/people.php/
D.2. TiN and Ti-O decoration of Single Wall Carbon Nanotubes and Graphene by Density Functional Theory computations
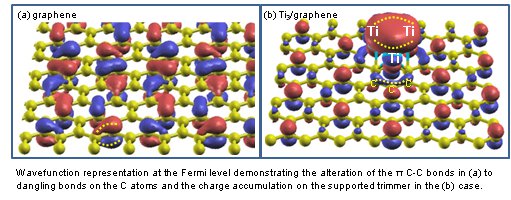
Ti nanostructures on Single Wall Carbon Nanotubes (SWCNTs) attracted considerable attention due to their
potential applications in electronic nanodevices and molecular adsorption. We report on Density Functional
Theory results referring to TiN (N=1,2,3,7,13) supported on SWCNTs and graphene. Two new
equivalent positions emerged that trisect the line joining the hexagon along the tube’s axis sides (TSH).
These sites accommodate dimmers and trimmers in compact linear and 2D triangular forms, respectively,
and 3D Ti7 and Ti13 conformations. Ti adsorbates introduce new electronic states
close and at the Fermi level. Despite the significant charge transfer from adsorbates to substrates,
these otherwise reduced TiN induce substantial charge screening in their surrounding substrate’s
ldlstoms and appear eventually as charged locations. These findings enlighten the early stages of Ti deposition,
predict possible active sites and may be of use for the design of metal-carbon coatings for applications
in catalysis and nano-electronics.
PhD student
Martha GialampoukiThesis: "Organic-inorganic hybrid nanomaterials with predetermined properties "
http://cmsl.materials.uoi.gr/people.php/
Publications
M.A. Gialampouki, Ch.E. Lekka
Journal of Physical Chemistry C 115 (2011) 15172-15181.
DOI: 10.1021/jp202130g
M.A. Gialampouki, M.A. Balerba, Ch.E. Lekka
Materials Chemistry and Physics 134 (2012) 214
DOI: journal_link
M.A. Gialampouki, Ch.E. Lekka
submitted
DOI: journal_link
.
2C. Bulk metallic glasses
Clustering and mechanical properties in Cu/Zr-based glassy models
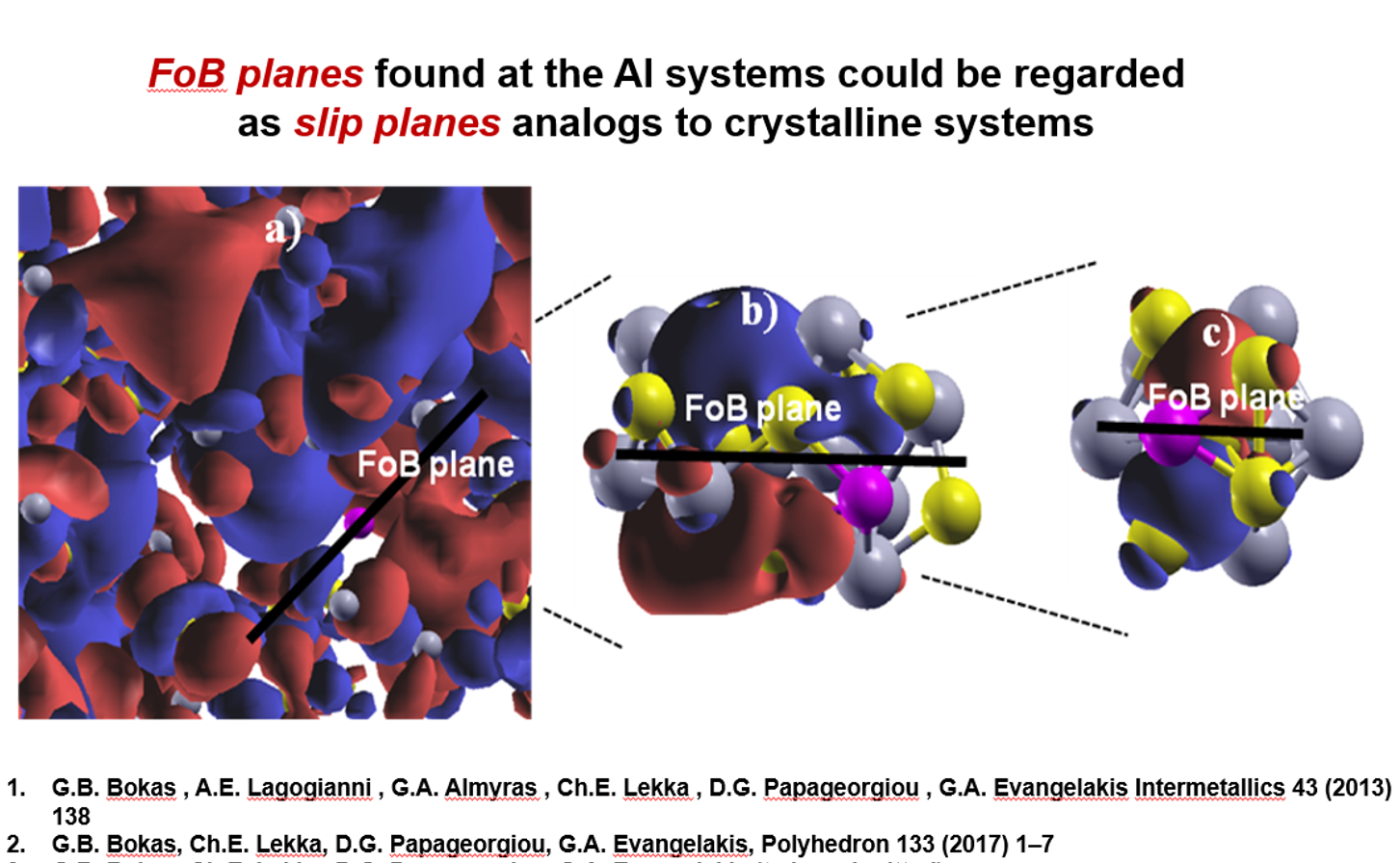
It is now accepted that Cu-Zr Metallic Glasses (MG) are mainly composed by small Icosahedral-like (ICO)
clusters that may be interconnected and/or interpenetrating. Using Molecular Dynamics and Reverse Monte
Carlo simulations we show that the interpenetrating ICOs carrying the compositional signature of the system
form SuperClusters (SC) which obey particular sequences of magic numbers. It comes out that these SCs are
reproducing very well the structural features of the experimental Radial Distribution Functions (RDF).
Interestingly, we found that the atomic pair distances do not depend on the respective compositions
of the systems, suggesting that the experimental shift of the RDFs when changing the stoichiometry
is due only to the alterations of the relative heights of the peaks. Upon deformation, the ICO clusters
are destructed and recreated while similar behavior follow the SCs explaining the way the Cu-Zr MGs
accommodate the stress upon deformation. In addition, we addressed the issue of Microalloying in
CuxZr12-xY (Y=Mg,Be,Al,Si,P,Nb,Ag) ICO cases and in selective CuZrAl and CuZrNb SCs by means
of Density Functional Theory calculations. We found significant alterations in the electronic
structures of the CuZr clusters and SCs manifested by new low-energy states and charge transfer
near the Fermi level. In the CuZrAl ICO and SC cases these new states are due to covalent-like
bonding between Al core and Cu shell atoms and Cu-Al core-core atoms, respectively. In the case
of Nb substitution the effects in the electronic structure are similar, but the interactions between
Nb core and Cu/Zr shell atoms are characterized by directional π-like bonds. In addition, we found
that a p-electron type dopant, as central atom, results in the creation of a plane with free of
core–shell atomic bonds, at certain energies and weak interactions at the Fermi level,
which could be viewed as a slip plane. s or d-electron type dopants may behave similarly due to
significant charge transfer towards unoccupied p-electrons occurring upon alloying. These
results could explain the experimental findings referring to the short range order, the
elucidation of the micro-alloying effect and the modifications induced in the mechanical
properties of these MGs by small Al or Nb additions.
Collaboration: G.A. Evangelakis, D.G. Papageorgiou
H. DIVERSITY: Improving the gender diversity management
in materials research institutions
Project : FP7-SIS-2008-1, No 230253
http://cordis.europa.eu/results/rcn/54518_en.html
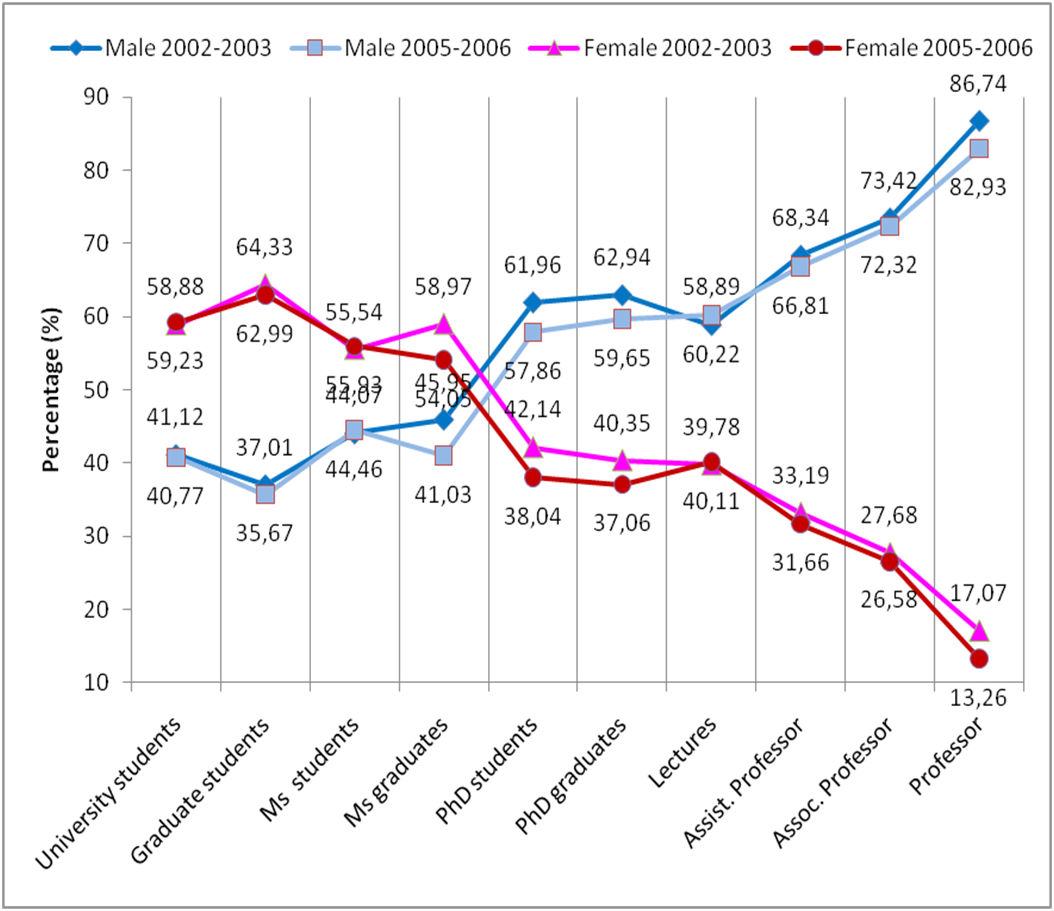
Figure: Percentage of male and female in different level of scientific career in Greece. "Gender Diversity from the Slovak and Greek Perspective" D. Caganova, O. Moravcik, J. Stefankova and M. Cambal, STU Slovakia and M. Gialampouki, Ch.E. Lekka UoI, Proceedings of the 22nd EAEEIE Annual Conference 2011 Maribor, Slovenia, June 13-15, 2011.